PHYLOGRAPHER OVERVIEW
PhyloGrapher is a program designed to visualize and study
evolutionary relationships within families of homologous
genes or proteins (elements). PhyloGrapher is a drawing tool
that generates custom graphs for a given set of elements.
In general, it is possible to use PhyloGrapher to visualize
any type of relations between elements.
Each gene or protein on PhyloGrapher's graph is represented
as a colored node (vertex) and connected to other nodes (vertices)
by lines (edges) of variable thickness and color based on the
similarity of genes or proteins (distance matrix). The position
of each node in the graph is flexible and adjusted by the user
to optimize visualization of the inter-relationships between the
nodes. Consequently, the physical distances on the graph between
nodes have no information content unlike classical phylogenetic
trees. The level of similarity between genes or proteins on
PhyloGrapher's graphs is indicated by color and line thickness.
PhyloGrapher is written in Tcl/Tk and works on Unix/Linux or Windows
that supports the Tcl/Tk toolkit which can be downloaded for free at
tcl.activestate.com. Macintosh
computers may have a problem running PhyloGrapher because of
the one-button mouse.
BASIC ELEMENTS and CHARACTERISTICS of GRAPHS
The critical characteristics of graphs in general are
which dots are connected by which lines.
The basic elements and characteristics of a graph are its:
vertex (node), edge, degree of a vertex (the number of edges
that touch it), size (number of vertices), path (a route from
vertex to vertex), length of the path (number of edges in a path),
planar and non-planar graphs (graph is planar if it can be drawn
on a plane so that the edges intersect only at the vertices),
distance (shortest path), diameter (longest distance between two
vertices), isolated vertex (a vertex of degree zero), adjacent
vertices (connected by an edge), neighborhood (adjacent vertices)
and others (you can check our
collection of web links).
A tree is a connected graph containing no cycles.
GOALS TO ACHIEVE WHEN MANIPULATING THE GRAPH
You may use a variety of criteria to maximize the readability
of the graph. In general, optimization goals can be:
Minimize: edge crossings and graph area;
Maximize: symmetries and smallest angle between edges.
You should pay attention to the basic elements of the graph, its
position and relationship (linkage), and make any kind of
scientific conclusion at your own risk. PhyloGrapher
is liscenced under the GPL and the author is not responsible
for anything that may happen.
INPUT FILES and PROGRAM OPERATION
To create a graph using PhyloGrapher you need to
set up two input files: 1. a
list of elements (genes or proteins) and
2. the distance matrix file.
Examine the files in the directory "Matrix".
The structure of these files is simple. "List" file contains
just IDs of elements (genes) of your data set. "Matrix" file
contains identity values for each pair of elements (genes).
PhyloGrapher reads data only from the first three columns of
"Matrix" file.
In the "Matrix" file identity values are in the third column and
they should be normalized between 0 and 1. All other columns in the
"Matrix" file are ignored by PhyloGrapher.
By clicking on "Load Data into Memory" the program reads the
list of genes and the matrix file and creates data structure in the
computer memory that will be used to construct
the graph on canvas by clicking on "Run". Nodes can be
assigned different colors representing different qualities
(e.g. species or linkage groups). You can paint nodes individually
or in "batch mode", click on "Node Painter" in "Extras".
PhyloGrapher initially generates non-organized graphs by placing
all elements in a circle in the same order as in the list of
elements in the input file and connecting related nodes by lines.
PhyloGrapher then allows you to move each node around using mouse
(click and drag) to make it easier to interpret the graph.
You can use the Canvas Editor from the "Extras" to finish editing
your graph. You can save coordinates of the nodes for future
projects. To do this, click on "Node Coords" under "Extras".
And finally, you can save your graph as postscript file, (or take a screenshot),
and if you want, generate HTML image map links for your graph.
PROGRAM FUNCTIONALITY and
KEY BINDINGS TO MANIPULATE THE IMAGE
Mouse:
left button - drag the node,
middle button - print node ID or edge weight on canvas
(the same as double left click in the Windows version),
right button - shake the node and view the degree.
Keyboard:
w - change color of node to white,
b - change color of node to blue,
s - change color of node to dark blue,
c - change color of node to cyan,
g - change color of node to green,
o - change color of node to orange,
r - change color of node to red,
v - change color of node to violet,
p - change color of node to purple.
Control-d - delete object from canvas
(You can not delete nodes and edges generated
from data file).
To find the node with a given ID, type the ID in the Node Entry
window and press Enter.
You can change the default font size of the printed ID and edge weight
by opening the "Canvas Editor" from "Extras" and changing tge font
size value in the corresponding window.
You can use PhyloGrapher to draw custom graphs by switching into
"Manual Mode". Choose from "Project Configuration" window
"Manual Drawing" option and click "Run". The empty canvas should
appear and using key "n" you can create new node. To draw the edge
from one node to another point mouse over the first node and press
"1" key, then point mouse over the second node and press "2" key.
New edge should appear.
GRAPHICAL FASTA/SSEARCH VIEWER
PhyloGrapher is highly integrated with
Bill Pearson's FASTA/SSEARCH programs. You can run FASTA/SSEARCH
in real time within PhyloGrapher for a given pair of sequences
(nodes) (FASTA and SSEARCH must be installed on your computer,
see ftp.virginia.edu/pub/fasta).
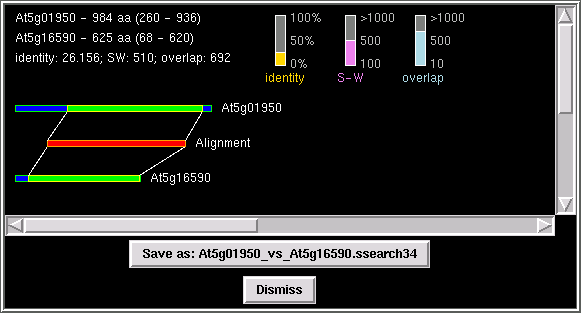
This feature allows the user to check different alignment parameters,
such as Smith-Waterman score, identity value, and overlap length.
PhyloGrapher runs FASTA/SSEARCH and parses the results of the search
and represents all data graphically
which simplify the validation of a given alignment.
To run FASTA/SSEARCH within PhyloGrapher:
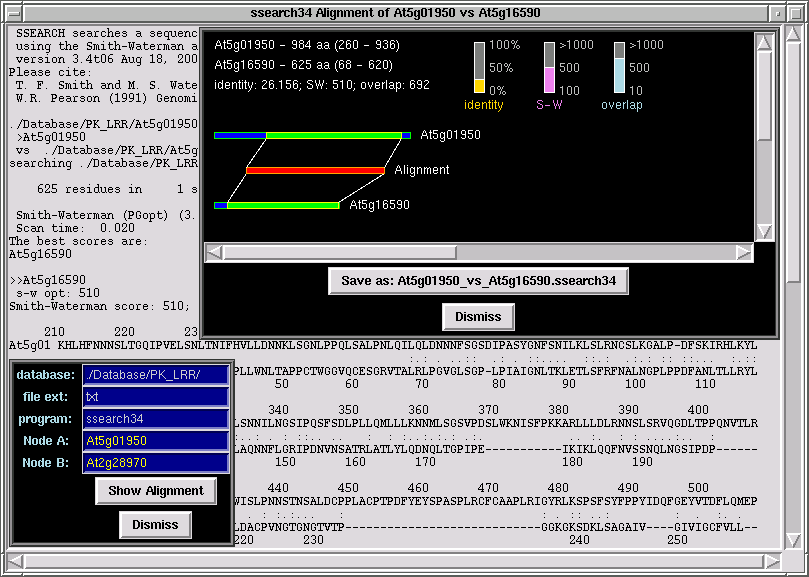
From the "Extras" menu click on "Smith-Waterman".
A new window will appear with following entry fields:
1. Database (directory where you store sequences in fasta format)
2. file extension (type of file extension of sequence files)
3. program (type of search to perform: SSEARCH by default or FASTA)
4. Node A ("query" sequence file)
5. Node B ("library" sequence file)
Point your mouse cursor over the "query" node (gene) and press <Control-a>;
the ID of node "A" in the corresponding entry window should appear. Then point
the mouse cursor over the "library" node (gene) and press <Control-s>;
the ID of node "B" in the corresponding entry window should appear. Then
click on "Show Alignment" and PhyloGrapher will run FASTA or SSEARCH
with a pair of sequences of your choice (node "A" vs node "B") and
display the graphical representation of the alignment.
You can save the results of the search as a text file as well as a postscript
file in the directory "Saved_Work" by clicking on the "save as" button.
You can change the font size of fasta output using the "Canvas Editor".
To install the FASTA and SSEARCH programs on your computer go to
Bill Pearson's ftp site
(ftp.virginia.edu/pub/fasta)
and download the current version of the FASTA distribution corresponding
to your computer platform.
On Windows uncompress the .zip FASTA file and copy
the executables fasta34.exe and ssearch34.exe into
the main WINDOWS directory. Read the FASTA documentation.
On Linux you need to compile the FASTA source code to get
the executables fasta34 and ssearch34. For that, copy
Makefile.linux to Makefile, and run "make".
Copy the fasta34 and ssearch34 binaries into the /usr/local/bin/ directory.
Read the FASTA documentation.
GRAPH TRAVERSAL (GRAPH SEARCH)
PhyloGrapher has a graph traversal (graph
search) functionality. For example, you can highlight
all nodes belonging to a single group (connected graph), or
you can select adjacent nodes to any given node. From the "Extras" dialog,
select "Adjacent nodes" and then generate the adjacency list file
by clicking on "Build Adjacency List". The adjacency list is based
on the data of the "list" and "matrix" files from main menu. Remember,
with different identity cutoff values ("no lines below...")
from main menu PhyloGrapher generates different adjacency lists.
As soon as the adjacency list is available you can select the node
you want to analyze by pointing the mouse cursor over the node.
Then click on "Highlight Adjacent Nodes" to highlight adjacent
nodes or "Highlight All Nodes in Group" to select all nodes
belonging to a connected graph. In this case PhyloGrapher
performs Depth-First Search (DFS) and you can observe the
progress of search visually.
GRAPH SELF ORGANIZATION
We attempted to implement a modified version of the
Fruchterman-Rheingold algorithm to organize the graph layout in
automatic mode. From "Extras" choose "Fruchterman-Rheingold"
and try to run it (Self Organize button). Try running it with the default
data set (My_Matrix_File.txt) to get an impression of what
the current version can achieve. It is far from perfect, but nodes move in the right direction
and form proper groups. You can try different parameters
to generate different graph layouts.
EXAMPLE DATA
PhyloGrapher contains example data for three large gene families
of Arabidopsis thaliana: NB-ARC (nucleotide binding site containing proteins,
putative resistance genes), cytochrome P450 and putative protein kinases (PK-LRR).
Protein sequences for selected subsets you can find under the directory
"Database/TIGR". All sequences derived from TIGR database
(ftp.tigr.org/pub/data/a_thaliana/ath1/SEQUENCES)
on September 2002.
Because the annotation, prediction of exon-intron structure, is
dynamic, protein sequences in this release of PhyloGrapher
may not correspond to current state of these sequences at
TIGR or other databases. This set is chosen as an example set
and author is not responsible for its regular update.
For your own project you may want to create new directory under
directory "Database" and place new set of sequences there.
Default set of genes "My_ID_List.txt" is a set of Arabidopsis cytochrome
P450 putative proteins. Corresponding matrix "My_Matrix_File.txt"
was derived by parsing of the results of BLAST search. You can find
the parser under directory "Scripts".
Feedback and comments may be sent to Alexander Kozik,
email: akozik@atgc.org
PhyloGrapher is under the GNU General Public License
Copyright © 2001 University of California at Davis, Alexander Kozik
{kind=link}
{kind=link}