Codon
Usage
Pipeline based on BLASTX search
Alexander
Kozik and Richard Michelmore
University of California at Davis
This codon usage
pipeline is based on analysis of results of ungapped BLASTX search. The
pipeline is a set of Python/Tcl-Tk scripts which extracts open reading
frames (ORFs) from BLAST alignments generated by BLASTX search and
counts codon usage for query sequences as well as for subject sequences
and generates report in the form of tab-delimited files. This approach
is useful when we want to find and extract putative ORFs from a set of
ESTs (query) by comparing (aligning) it with a set of sequences with
known ORFs (subject in BLAST terminology). Dataflow of this pipeline is
shown below:
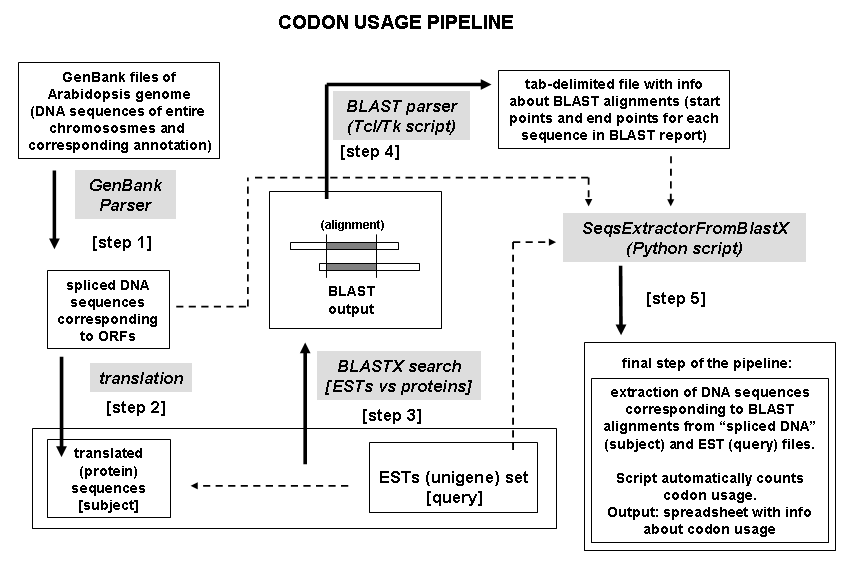
Input files:
1. "DNA"
file which corresponds to open reading
frames of the subject sequences
2.
"Protein" file which derived by translation of
"DNA" file
For example, Arabidopsis ORFs and their translated
sequences were derived from NCBI GenBank files using Python GenBank parser:
ath_NCBI_cds.fasta.nr.info.gz
(Arabidopsis CDS or "DNA" file)
ath_NCBI_genes.fasta.nr.info.gz
(corresponding protein sequences or "protein" file)
(Note, instead of Arabidopsis files user can
use any other dataset as long as "protein" file is derived from "DNA"
file)
3. cos_soybean.fasta
(example set of soybean ESTs or "Query" file for which we want to find
ORFs)
Programs
and scripts:
1. NCBI Blast program (you need to
download and install BLAST program from NCBI ftp site)
2. tcl_blast_parser_123_V025.tcl
(read more at: http://www.atgc.org/BlastParser/Blast_Parser_017.html)
3. SeqsExtractorFromBlastX_024_V07d.py
Pipeline
usage:
1. run BLASTX
search of cos_soybean.fasta (ESTs
query set) against Arabidopsis protein database:
> blastall -p blastx
-F "m
S" -g F -d ./ath_NCBI_genes.fasta.nr.info
-i ./cos_soybean.fasta -o
x-cos_soybean_vs_ath_24hits_no_gaps.out
-e 1e-10 -v 24 -b 24
after some time (1-2 hours on 1 GHz CPU) x-cos_soybean_vs_ath_24hits_no_gaps.out
will be generated
2. run
tcl_blast_parser on BLASTX output:
>
tclsh tcl_blast_parser_123_V025.tcl
x-cos_soybean_vs_ath_24hits_no_gaps.out
x-cos_soybean_vs_ath_24hits_no_gaps.out 20 40 100
MATRIX
after parsing several files will be generated (see
details at http://www.atgc.org/BlastParser/Blast_Parser_017.html).
We are interested in x-cos_soybean_vs_ath_24hits_no_gaps.out.info2
file only which contains following information:
- 1
column: "Query" sequence ID
- 2 column: "Subject" sequence ID
- 3 column: description line for "Subject" sequence
- 4 column: normalized expectation (-log(Exp)/100)
[note, that expectation is normalized between 0 and 1]
- 5 column: percent of identity
- 6 column: number of perfect matches
- 7 column: length of the alignment
- 8 column: hit number for primary alignment (1 is the
best first hit)
- 9 column: frame of translation (+1, +2, +3, -1, -2
or -3)
- 10 column: first position of the "Query" sequence in the
alignment
- 11 column: last position of the "Query" sequence in the
alignment
- 12 column: first position of the "Subject" sequence in the
alignment
- 13 column: last position of the "Subject" sequence in the
alignment
- 14 column: Length of "Query"(nt or aa) / Length of
"Subject"(nt or aa)
- 15 column: gap info
SeqsExtractorFromBlastX will use information
from "*.info2" file to extract sub-sequences from query and subject
3. run
SeqsExtractorFromBlastX script as described below:
>
python SeqsExtractorFromBlastX_024_V07d.py cos_soybean.fasta
ath_NCBI_cds.fasta.nr.info
x-cos_soybean_vs_ath_24hits_no_gaps.out.info2
x-cos_soybean.codons x-cos_soybean.align
SeqsExtractorFromBlastX_024_V07d.py script takes five
arguments as input:
- 1-st
argument:
cos_soybean.fasta
file contains EST
sequences (BLASTX query)
- 2-nd
argument:
ath_NCBI_cds.fasta.nr.info file contains DNA
sequences of BLASTX database (subject)
-
3-d argument:
x-cos_soybean_vs_ath_24hits_no_gaps.out.info2 file
- 4-th
argument:
x-cos_soybean.codons is output file(s) of
SeqsExtractorFromBlastX script
- 5-th
argument:
x-cos_soybean.align is a directory where
all extracted alignments will be written
SeqsExtractorFromBlastX
script will generate a new directory with DNA sequence alignments in
FASTA format:
x-cos_soybean.align
Each alignment is extracted according to first best
hit of BLASTX report and written into separate file. Also, each
alignment corresponds to open reading frames only and does not contain
stop codons within query or subject sequences. However alignments may
contain unambiguous letters (N or X, for example).
You can download compressed example output here: x-cos_soybean.align.tar.gz
Other output files of the SeqsExtractorFromBlastX
script:
- x-cos_soybean.codons.overlap_info
- contains positional information about extracted subsequences (e.g.
from which part of original sequence any given subsequence was
extracted)
- x-cos_soybean.codons.query_seq -
file with extracted "query" subsequences in FASTA format (fragments of
soybean ESTs corresponding to ORFs according to BLASTX alignments of
this example)
- x-cos_soybean.codons.subj_seq - file with extracted
"subject" subsequences in FASTA format (fragments of Arabidopsis ORFs
corresponding to BLASTX alignments of this example)
- x-cos_soybean.codons.triplets -
info file with codon usage for "query" and "subject" sequences (counts
all triplets)
- x-cos_soybean.codons.x_kska
- file with information about synonymous/non-synonymous substitutions
for every non-gapped alignment
- x-cos_soybean.codons.x_triplets
- info file with codon
usage for "query" and "subject" sequences (conservative case, e.g. this
file contains information for triplets only for cases when both
triplets (query and subject) are informative (do not contain
unambiguous letters).
Detailed output explanation:
Column description
of "*.triplets" and "*.x_triplets" files:
-
1-st column: amino acid
-
2-nd column: triplet
-
3-d column: "query" triplet usage per 100,000 cases [query_value]
-
4-th column: "subject" triplet usage per 100,000 cases [subject_value]
-
5-th column: [diff_value] difference between 3-d and 4-th columns (
[diff_value] = [query] minus [subject] )
-
6-th column: difference in percentages ( [diff_value%] = [diff_value] /
[subject_value] * 100 )
-
7-th column: total number of the given triplet [query]
-
8-th column: total number of all analyzed triplets [query]
-
9-th column: total number of the given triplet [subject]
-
10-th column: total number of all analyzed triplets [subject]
-
11-th column: percentage of query codon usage
-
12-th column: percentage of subject codon usage
-
13-th column: difference between column 11 and 12 ( [query] minus
[subject] )
-
14-th column "***" visual mark
-
15-th column: percentage of query amino acid usage
-
16-th column: percentage of subject amino acid usage
-
17-th column: difference between column 15 and 16 ( [query] minus
[subject] )
(Note,
that the total number of all analyzed triplets of query and the total
number of all analyzed triplets of subject may be different in the file
"*.triplets" and must be the
same in "*.x_triplets")
Column description
of "*x_kska" file:
-
1-st column: query ID
-
2-nd column: subject ID
-
3-d column: number of synonymous substitutions
-
4-th column: number of non-synonymous substitutions
-
5-th column: difference between columns 3 and 4
-
6-th column: "***" visual mark
-
7-th column: number of perfect codon matches
-
8-th column: number of amino acid matches
-
9-th column: total number of triplets in the alignment
-
10-th column: "***" visual mark
-
11-th column: identity (%) at protein level
-
12-th column: ratio of [perfect codon
matches] to [amino acid matches] ((column7 /
column8) * 100)
(Note, that the sum of values in columns 3 and 7 must be equal to value
of column 8 ([synonymous substitutions] plus [perfect codon matches] =
[amino acid matches])
Additional
scripts to determine GC% content in a set of sequences:
Finally, we can calculate GC% content for our extracted EST
query ORFs subset of sequences using seqs_processor_017_u.py
script:
> python seqs_processor_017_u.py x-cos_soybean.codons.query_seq
x-cos_soybean.codons.query_seq.proc DNA
Script takes three arguments ([input], [output],
["DNA" option]) and generates three output files:
We
are interested in x-cos_soybean.codons.query_seq.proc.stat file which contains
information about GC% content for every sequence as well as summary
(last line)
(Note, seqs_processor_017_u.py script will terminate
if gene ID duplication takes place in the input file. seqs_processor_017_d.py should work
with duplicated IDs)
py_stat_graph_007.py script can
process x-cos_soybean.codons.query_seq.proc.stat
file and generate MS excel ready output to show distribution of DNA
sequences proportional to their GC% content:
>
python py_stat_graph_007.py x-cos_soybean.codons.query_seq.proc.stat
x-cos_soybean.codons.query_seq.proc.stat.graph 1 100
8
py_stat_graph_007.py takes five
arguments ( [input] [output] [min_value] [max_value] [column_number] ),
analyzes defined column of input file and generates statistical
information: x-cos_soybean.codons.query_seq.proc.stat.graph.
click here
to find out how to obtain multiple alignments of orthologs by combining two subsets
generated by pipeline described above.
DNA translation in six frames Python script
email: akozik@atgc.org
last modified: April 21 2006