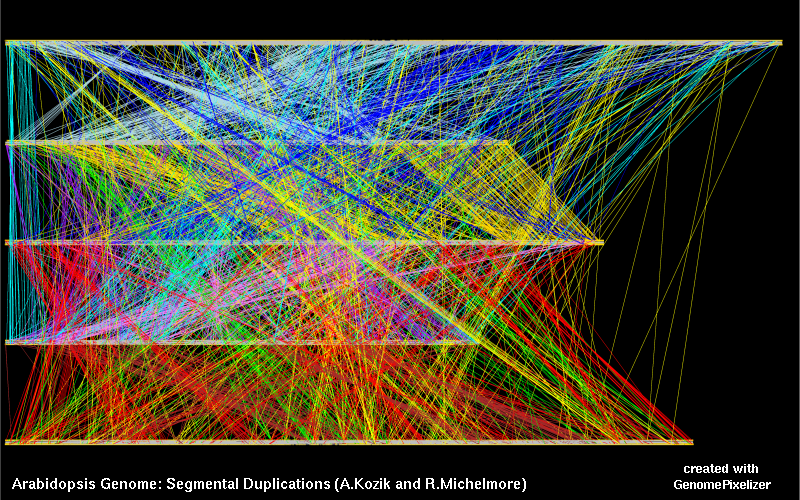
Processing of Arabidopsis GenBank files
Current version of
Arabidopsis GenBank files
contains information about genes
with alternative splicing variants. It means that the same gene can be represented
several times depending on how many splicing variants it has. If FASTA file with
protein sequences derived from such GenBank file contains sequences with splicing
variants it may affect results of computational analysis. For example, if we are
looking for the set of single copy genes in Arabidopsis genome by BLAST-ing of
"everything against everything" and if a single copy gene has alternative splicing
variants in the file we analyze then we can easily overlook this gene because of
multiple hits to itself. Also, if we are using file containing splicing variants
to display gene distribution over chromosomes by GenomePixelizer then the same
gene can be displayed wrongly several times showing false illusion of gene clustering.
It was not a big problem long time ago when information about alternative splicing
in Arabidopsis was rare. Current version of annotation of Arabidopsis genome contains
more than one thousand alternative spliced genes. For that reason FASTA file and
file with gene coordinates derived from GenBank files has to be pre-processed to
remove all redundant sequence IDs, so every gene is represented just one time
regardless of the number of alternative splicing variants. It is hard to choose
which splice variant to pick up. It can be the longest variant, or variant based
on the number of supporting ESTs, or just a first variant (CDS entry) how it is
listed in GenBank file.
On this web page you can download:
to use them with GenomePixelizer or GenoPix2D plotter.
FASTA file with predicted protein sequences and gene coordinates were extracted
from NCBI Arabidopsis GenBank files
(Release 4.0, May 14, 2003) using
Python GenBank parser.
Sequences with alternative splicing variants were removed by custom scripts.
Only first CDS for cases with alternative splicing remained. Next version
of our GenBank parser will do it automatically.
Matrix file was generated from BLAST output using
tcl_blast_parser
with default options. BLAST options were:
./blastall -p blastp -F F -d ./ath_ncbi_Jul_2003.fasta -i ./ath_ncbi_Jul_2003.fasta
-o ath_vs_ath_TIGR_2003_July_240hits.out -e 1e-20 -v 240 -b 240 -I T &
Current distributions of GenomePixelizer and GenoPix2D plotter contain
data for old Arabidopsis Release 3.0. You can update input files to Release 4.0 by
copying of ath_ncbi_Jul_2003.coords, ath_ncbi_Jul_2003.matrix and
ath_ncbi_Jul_2003.annotation into "Arabidopsis_Genome" directory in the case of
GenomePixelizer, and "Matrix" directory in the case of GenoPix2D plotter.
Minor modification of GenomePixelizer RunSetup file is required (change input
file names in options 1, 2 and 19). Also, you need to re-run
DiagHunter program
to find duplicated genome regions in the case of GenoPix2D plotter.
Also, you may want to re-run
hmmsearch on ath_ncbi_Jul_2003.fasta file to find
Pfam domains for new Arabidopsis annotation release.
HMM models for NBS, P450, LRR and pkinase can be found under directory "Misc" of
GenoPix2D plotter program.
|